Apoptosis or necroptosis? The caspase-8 protein decides

Doug Green, PhD, left, and Bart Tummers, PhD, write about a protein that determines the fate of cells.
A broker of cell death
Cells kill themselves on purpose all of the time. This is usually a good thing, as it clears out sick or unnecessary cells from the body and helps avoid disease.
But cell death is not a simple event. Many triggers can cause it, including stress, infections and pro-cancer signals. And there are different ways for cells to die. Excessive damage can kill a cell, but cells can also decide to die on their own accord. This is stimulated by signals from within or without and is regulated by sophisticated molecular mechanisms; the death is ‘programmed.’ Infections and pro-cancer signals produce extrinsic factors that can initiate two types of programmed cell death: apoptosis and necroptosis. If and how these extrinsic factors kill the cell hinges on an intricate interplay between molecular events.
Caspase-8: Executioner, Protector
One of the central decision-makers is the protein caspase-8. Caspase-8 gains the ability to cleave other proteins when the cell is activated by extrinsic signals. It then processes a cell death executioner protein, named caspase-3, and the cell dies by apoptosis.
However, caspase-8 can also protect cells from death. For this it relies on the expression of the protein cFLIPL. Intricately, cFLIPL expression is induced by the same extracellular signals that govern death. Expressed cFLIPL stops the ability of caspase-8 to induce apoptosis and the cell lives. The extracellular signals induce a third event; activation of the proteins RIPK1, RIPK3 and MLKL. This results in death of the cell by necroptosis.
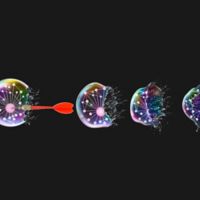
Cells that die by apoptosis "bleb" right before they die.
Cells dying by necroptosis round up and "pop," releasing their content in the surrounding milieu.
cFLIPL directs caspase-8 to cleave RIPK1 and RIPK3 and this stops necroptosis. The expression of cFLIPL thus protects the cell from both apoptotic and necroptotic cell death and simultaneously allows the expression of factors that cause inflammation.
What hangs in the balance?
The type of death that is chosen can have major effects on the body's immune response to cancer and inflammatory disease.. Apoptosis is generally a silent mode of cell death, the surrounding cells or the immune system are not alarmed. But necroptotic cells can alarm surrounding cells, and this can lead to massive inflammation.
Why do cells have this balance? Why are cFLIPL and caspase-8 important? Some pathogens can interfere with the expression of cFLIPL. This can activate caspase-8 and the cell dies by apoptosis. Other pathogens interfere with the function of caspase-8. Caspase-8 then no longer can block RIPK1 and RIPK3 and the balance tips toward necroptosis. This kills the infected cell, taking away the ‘home’ of the pathogen, warns surrounding cells and stimulates the attraction of immune cells that can form the defense against the pathogen. So far so good. But what happens when pathogens interfere with necroptosis? Does the body then stay infected? Fortunately not.
Cells have other ways of signaling to the immune system that there is an infection. And the immune system will clear the infection. However, when cancers eliminate the expression of RIPK3 they also are protected from necroptosis. Immune cells are trained not to attack cells that come from the body, so cancer cells will not be cleared by the immune system. Many cancer types use this strategy of reducing RIPK3 expression to escape necroptosis. An option could be to inhibit the expression of cFLIPL, so that the cancer cells could die by apoptosis. Unfortunately, this gives severe side effects, because healthy cells are also affected. Other strategies thus need to be developed. And this requires an even deeper understanding of how these intricate molecular mechanisms are regulated. The identification of other molecules, interactions or therapies may lead to an innovative strategy for the treatment of cancer.
In a recent review article in Immunological Reviews, we take a close look at the complex interplay between caspase-8, FADD and cFLIP in different types of cell death and inflammation. Read the review article.
About the author